Tecentriq (Atezolizumab) – Rezumatul caracteristicilor produsului
Acest medicament – Tecentriq (Atezolizumab) – Rezumatul caracteristicilor produsuluiface obiectul unei monitorizări suplimentare. Acest lucru va permite identificarea rapidă de noi informaţii referitoare la siguranţă. Profesioniştii din domeniul sănătăţii sunt rugaţi să raporteze orice reacţii adverse suspectate. Vezi pct. 4.8 pentru modul de raportare a reacţiilor adverse.
Prospect Tecentriq (Atezolizumab)
Prospectul il puteti gasi aici, mai jos gasiti rezumatul caracteristicilor produsului.
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
- 4. DATE CLINICE
- 4.1 Indicaţii terapeutice
- 4.2 Doze şi mod de administrare
- 4.3 Contraindicaţii
- 4.4 Atenţionări şi precauţii speciale pentru utilizare
- 4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
- 4.6 Fertilitatea, sarcina şi alăptarea
- 4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
- 5. PROPRIETĂŢI FARMACOLOGICE
- 6. PROPRIETĂŢI FARMACEUTICE
- 7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
- 10. DATA REVIZUIRII TEXTULUI
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
Tecentriq 1200 mg concentrat pentru soluţie perfuzabilă
2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
Un flacon a 20 ml concentrat conţine atezolizumab 1200 mg *.
După diluare (vezi pct. 6.6), un ml de soluţie conţine atezolizumab aproximativ 4,4 mg.
*Atezolizumab este un anticorp monoclonal umanizat de tip IgG1 cu acţiune împotriva lingandului 1 cu rol în controlul morţii celulare programate (PD-L1) modificat la nivelul regiunii Fc, produs în celule ovariene de hamster chinezesc prin tehnologie ADN recombinant.
Pentru lista tuturor excipienţilor, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Concentrat pentru soluţie perfuzabilă
Lichid limpede, incolor până la galben pal
4. DATE CLINICE
4.1 Indicaţii terapeutice
Tecentriq în monoterapie este indicat pentru tratamentul carcinomului urotelial (CU) local avansat sau metastazat, la pacienţi adulţi, după tratament anterior cu chimioterapie cu săruri de platină sau care nu sunt consideraţi eligibili pentru tratamentul cu cisplatină, vezi pct. 5.1.
Tecentriq în monoterapie este indicat pentru tratamentul neoplasmului bronho-pulmonar altul decât cel cu celule mici (NSCLC) local avansat sau metastazat, la pacienţi adulţi, după tratament anterior cu chimioterapie. Pacienților cu mutaţii activatoare ale EGFR sau mutaţii tumorale ALK pozitive trebuie, deasemenea, să li se fi administrat tratament specific, înaintea administrării Tecentriq (vezi pct. 5.1).
4.2 Doze şi mod de administrare
Tratamentul cu Tecentriq trebuie iniţiat şi supravegheat de medici cu experienţă în tratamentul cancerului.
Doze
Doza recomandată de Tecentriq este de 1200 mg, administrată prin perfuzie intravenoasă la interval de trei săptămâni.
Durata tratamentului
Se recomandă ca pacienţii să fie trataţi cu Tecentriq până la pierderea beneficiului clinic (vezi pct. 5.1) sau până când toxicitatea devine imposibil de gestionat.
Doze întârziate sau omise
Dacă o doză de Tecentriq planificată este omisă, aceasta trebuie administrată cât mai curând posibil; nu se aşteaptă până la următoarea doză planificată. Planificarea administrării trebuie modificată pentru a menţine un interval de 3 săptămâni între doze.
Modificările dozei pe durata tratamentului
Nu se recomandă reduceri ale dozei de Tecentriq.
Doze întârziate sau întreruperea administrării dozei (vezi, de asemenea, pct. 4.4 şi 4.8)
Tabelul 1: Recomandări privind modificarea dozei pentru reacţii adverse specificate la
medicament
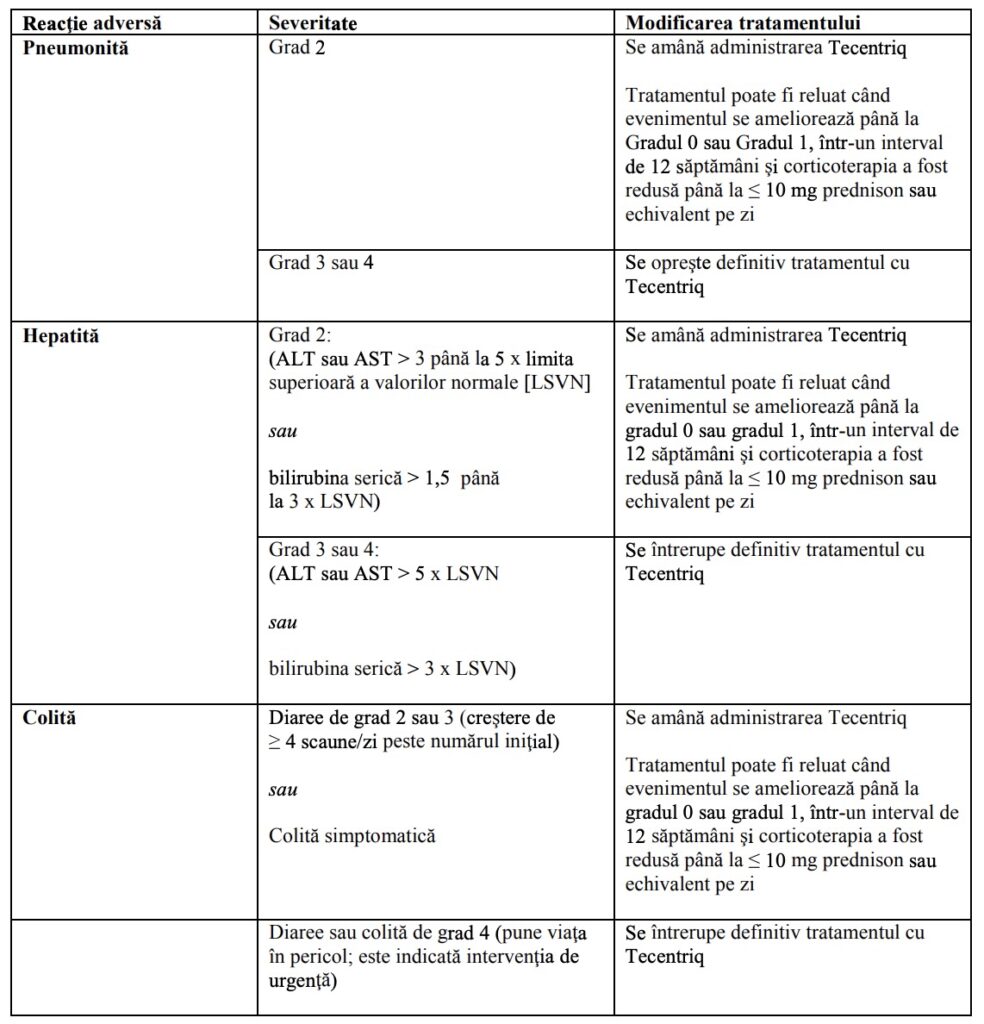
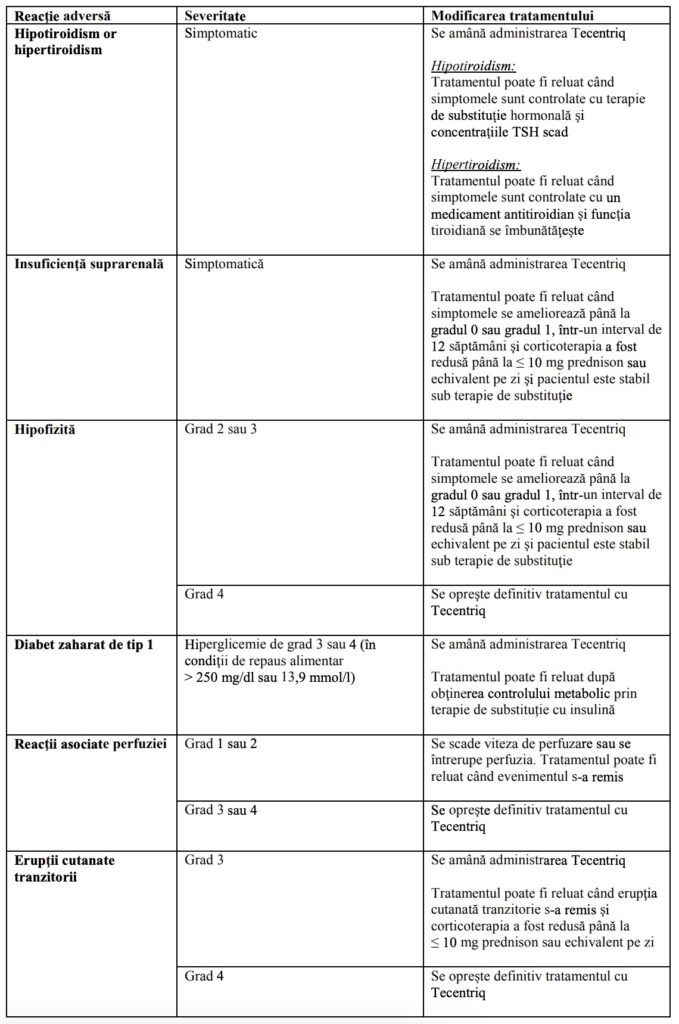
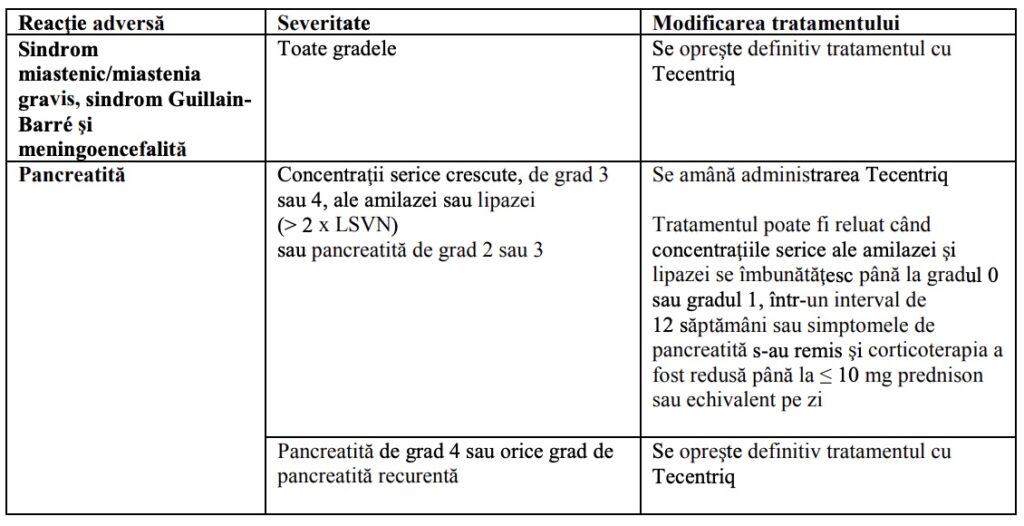
Notă: Gradele de toxicitate sunt în conformitate cu Criteriile de Terminologie Comună pentru Evenimente Adverse ale Institutului Naţional de Cancer versiunea 4.0 (NCI-CTCAE v4).
Tratamentul cu Tecentriq trebuie întrerupt definitiv:
- În cazul toxicităţilor de grad 4, cu excepţia endocrinopatiilor care sunt controlate prin tratament de substituţie hormonală
- În cazul recurenţei oricărui eveniment cu grad de severitate ≥ 3
- În cazul în care toxicitatea asociată tratamentului nu se ameliorează până la grad 0 sau grad 1 în decurs de 12 săptămâni de la debutul reacţiei adverse.
- În cazul în care este necesară corticoterapie în doză de > 10 mg prednison sau echivalent pe zi pentru tratamentul toxicităţii asociate după mai mult de 12 săptămâni de la debutul reacţiei adverse
Pacienţilor trataţi cu Tecentriq trebuie să li se înmâneze Cardul de avertizare pentru pacient şi să li se aducă la cunoştinţă riscurile administrării Tecentriq (vezi, de asemenea, prospectul).
Grupe speciale de pacienţi
Copii şi adolescenţi
Siguranţa şi eficacitatea Tecentriq la copii şi adolescenţi cu vârsta sub 18 ani nu au fost încă stabilite. Nu sunt disponibile date.
Pacienţi vârstnici
Pe baza unei analize de farmacocinetică populaţională, nu este necesară ajustarea dozelor de Tecentriq la pacienţii cu vârsta ≥ 65 ani.
Insuficienţă renală
Pe baza unei analize de farmacocinetică populaţională, nu este necesară ajustarea dozei la pacienţi cu insuficienţă renală uşoară sau moderată (vezi pct. 5.2). Datele provenite de la pacienţi cu insuficienţă renală severă sunt prea limitate pentru a permite formularea unor concluzii referitoare la această grupă de pacienţi.
Insuficienţă hepatică
Pe baza unei analize de farmacocinetică populaţională, nu este necesară ajustarea dozei la pacienţi cu insuficienţă hepatică uşoară. Tecentriq nu a fost studiat la pacienţi cu insuficienţă hepatică moderată sau severă (vezi pct. 5.2).
Statusul de performanţă Eastern Cooperative Oncology Group (ECOG) ≥ 2
Pacienţii cu status de performanţă ECOG ≥ 2 au fost excluşi din studiile clinice efectuate pentru indicaţia de NSCLC şi din studiile cu indicaţie de terapie de linia a 2-a pentru CU (vezi pct. 4.4 şi 5.1).
Mod de administrare
Tecentriq este destinat administrării intravenoase. Perfuzia nu trebuie administrată intravenos rapid sau în bolus intravenos.
Doza iniţială de Tecentriq trebuie administrată pe durata a 60 minute. Dacă prima perfuzie este bine tolerată, toate perfuziile ulterioare pot fi administrate pe durata a 30 minute.
Pentru instrucţiuni privind diluarea şi manipularea medicamentului înainte de administrare, vezi pct. 6.6.
4.3 Contraindicaţii
Hipersensibilitate la atezolizumab sau la oricare dintre excipienţii enumeraţi la pct. 6.1.
4.4 Atenţionări şi precauţii speciale pentru utilizare
În scopul îmbunătăţirii trasabilităţii medicamentelor biologice, denumirea comercială şi seria de fabricaţie a medicamentului administrat trebuie înregistrată clar (sau menţionată) în fişa pacientului.
Majoritatea reacţiilor adverse mediate imun care au apărut pe parcursul tratamentului cu atezolizumab au fost reversibile şi abordate terapeutic prin întreruperea tratamentului cu atezolizumab şi iniţierea corticoterapiei şi/sau tratamentului de susţinere. Au fost observate reacţii adverse mediate imun care afectează mai mult de un aparat sau sistem.
Reacţiile adverse mediate imun induse de atezolizumab pot apărea după administrarea ultimei doze de atezoliumab.
În cazul reacţiilor adverse suspectate a fi mediate imun, trebuie efectuată o evaluare completă pentru a confirma etiologia sau a exclude alte cauze. În funcţie de gradul de severitate a reacţiei adverse, administrarea atezolizumab trebuie amânată şi trebuie administrată corticoterapie. După ameliorare până la gradul ≤ 1, corticoterapia trebuie scăzută treptat pe parcursul unei perioade de cel puţin 1 lună.
Pe baza datelor limitate provenite din studiile clinice efectuate la pacienţi ale căror reacţii adverse mediate imun nu au putut fi controlate prin corticosterapie sistemică, poate fi luată în considerare administrarea altor imunosupresoare sistemice.
Tratamentul cu atezolizumab trebuie întrerupt definitiv în cazul recurenţei oricărei reacţii adverse mediate imun de grad 3 şi în cazul oricărei reacţii adverse mediate imun de grad 4, cu excepţia endocrinopatiilor care sunt controlate cu tratament de substituţie hormonală (vezi pct. 4.2 şi 4.8).
Pneumonită mediată-imun
În studiile clinice, s-au observat cazuri de pneumonită, inclusiv cazuri letale, în asociere cu
atezolizumab (vezi pct. 4.8). Pacienţii trebuie monitorizaţi pentru apariţia semnelor şi simptomelor de pneumonită.
Tratamentul cu atezolizumab trebuie amânat în cazul pneumonitei de grad 2 şi trebuie iniţiat tratamentul cu 1-2 mg/kg şi zi de prednison sau un echivalent. Dacă simptomele se ameliorează până la ≤ gradul 1, corticoterapia trebuie scăzută treptat pe parcursul a ≥ 1 lună. Tratamentul cu atezolizumab poate fi reluat dacă evenimentul se ameliorează până la grad ≤ 1 într-un interval de 12 săptămâni şi dacă doza de corticosteroizi a fost redusă la ≤ 10 mg prednison sau un echivalent pe zi.
Tratamentul cu atezolizumab trebuie întrerupt definitiv în cazul pneumonitei de grad 3 sau 4.
Hepatită mediată-imun
În studiile clinice, s-au observat cazuri de hepatită, unele dintre acestea cu consecinţe letale, înasociere cu atezolizumab (vezi pct. 4.8). Pacienţii trebuie monitorizaţi pentru apariţia semnelor şi simptomelor de hepatită.
Aspartat aminotransferaza (AST), alanin aminotransferaza (ALT) şi bilirubina trebuie monitorizate înainte de începerea tratamentului, periodic pe durata tratamentului cu atezolizumab şi după cum este indicat pe baza evaluării clinice.
Tratamentul cu atezolizumab trebuie amânat dacă valorile crescute ale evenimentelor de grad 2 (valorile serice ale ALT sau AST > 3 până la 5 x LSVN sau ale bilirubinei > 1,5 până 3 x LSVN) persistă mai mult de 5 până la 7 zile şi trebuie iniţiat tratamentul cu prednison în doză de 1 până la 2 mg/kg pe zi sau echivalent. În cazul în care evenimentul se ameliorează până la un grad ≤1, corticoterapia trebuie scăzută treptat pe parcursul a ≥ 1 lună.
Tratamentul cu atezolizumab poate fi reluat dacă evenimentul se ameliorează până la grad ≤ 1 într-un interval de 12 săptămâni şi corticoterapia a fost redusă până la ≤ 10 mg prednison sau un echivalent pe zi. Tratamentul cu atezolizumab trebuie întrerupt definitiv în cazul evenimentelor de grad 3 sau grad 4 (valorile serice ale ALT sau AST > 5,0 x LSVN sau ale bilirubinei > 3 x LSVN).
Colită mediată-imun
În studiile clinice s-au observat cazuri de diaree sau colită în asociere cu atezolizumab (vezi pct. 4.8).
Pacienţii trebuie monitorizaţi pentru apariţia semnelor şi simptomelor de colită.
Tratamentul cu atezolizumab trebuie amânat în cazul diareei de grad 2 sau 3 (creştere de ≥ 4 scaune/zi peste numărul iniţial) sau al colitei (simptomatică). Trebuie iniţiat tratamentul cu prednison în doză de 1-2 mg/kg şi zi sau echivalent în cazul diareei sau colitei de grad 2, dacă simptomele persistă > 5 zile sau dacă reapar. Trebuie iniţiată corticoterapia administrată intravenos (1-2 mg/kg şi zi de metilprednisolon sau echivalent) în cazul diareei sau colitei de grad 3. Imediat ce simptomele se ameliorează, trebuie început tratamentul cu prednison în doză de 1-2 mg/kg şi zi sau echivalent. Dacă
simptomele se ameliorează la un grad ≤ 1, corticoterapia trebuie scăzută treptat pe parcursul a ≥ 1 lună.
Tratamentul cu atezolizumab poate fi reluat dacă evenimentul se ameliorează până la un grad ≤ 1 întrun interval de 12 săptămâni şi corticoterapia a fost redusă până la ≤ 10 mg prednison sau echivalent pe zi. Tratamentul cu atezolizumab trebuie întrerupt definitiv în cazul diareei sau colitei de grad 4 (pune viaţa în pericol; este indicată intervenţie de urgenţă).
Endocrinopatii mediate-imun
În studiile clinice s-au observat hipotiroidism, hipertiroidism, insuficienţă suprarenală, hipofizită şi diabet zaharat de tip 1, incluzând cetoacidoză diabetică, în asociere cu atezolizumab (vezi pct. 4.8).
Pacienţii trebuie monitorizaţi pentru apariţia semnelor şi simptomelor de endocrinopatii. Funcţia tiroidiană trebuie monitorizată înainte de iniţierea şi periodic pe durata tratamentului cu atezolizumab.
Trebuie luată în considerare conduita terapeutică adecvată în cazul pacienţilor cu valori anormale ale testelor funcţiei tiroidiene la momentul iniţial.
Pacienţii asimptomatici care au valori anormale ale testelor funcţiei tiroidiene pot fi trataţi cu atezolizumab. În cazul hipotiroidismului simptomatic, trebuie amânată administrarea atezolizumab şi trebuie iniţiată terapia de substituţie cu hormoni tiroidieni, după cum este necesar. Hipotiroidismul izolat poate fi tratat cu terapie de substituţie, fără corticoterapie. În cazul hipertiroidismului simptomatic, trebuie amânată administrarea atezolizumab şi trebuie iniţiat tratamentul cu un medicament antitiroidian, după cum este necesar. Tratamentul cu atezolizumab poate fi reluat atunci când simptomele sunt controlate şi funcţia tiroidiană se ameliorează.
În cazul insuficienţei suprarenale simptomatice, trebuie amânată administrarea atezolizumab şi trebuie iniţiată corticoterapia administrată intravenos (1-2 mg/kg şi zi de metilprednisolon sau echivalent).
Imediat ce simptomele se ameliorează, trebuie instituit tratamentul cu prednison în doză de 1-2 mg/kg şi zi sau echivalent. Dacă simptomele se ameliorează până la grad ≤ 1, corticoterapia trebuie scăzută treptat pe parcursul a ≥ 1 lună. Tratamentul poate fi reluat dacă evenimentul se ameliorează până la grad ≤ 1 într-un interval de 12 săptămâni şi corticoterapia a fost redusă până la ≤ 10 mg prednison sau echivalent pe zi şi pacientul este stabil sub terapie de substituţie (dacă este necesară).
În cazul hipofizitei de grad 2 sau de grad 3, trebuie amânată administrarea atezolizumab şi trebuie iniţiată corticoterapia pe cale intravenoasă (1-2 mg/kg şi zi metilprednisolon sau echivalent), precum şi tratamentul de substituţie hormonală, după cum este necesar. Imediat ce simptomele se ameliorează, trebuie instituit tratamentul cu prednison în doză de 1-2 mg/kg şi zi sau echivalent. Dacă simptomele se ameliorează până la grad ≤ 1, corticoterapia trebuie scăzută treptat pe parcursul a ≥ 1 lună.
Tratamentul poate fi reluat dacă evenimentul se ameliorează până la grad ≤ 1 pe parcursul a 12 săptămâni şi corticoterapia a fost redusă până la ≤ 10 mg prednison sau echivalent pe zi şi pacientul este stabil sub terapie de substituţie (dacă este necesară). Tratamentul cu atezolizumab trebuie întrerupt definitiv în cazul hipofizitei de grad 4.
Trebuie iniţiat tratamentul cu insulină pentru diabetul zaharat de tip 1. În cazul hiperglicemiei de grad ≥ 3 (glicemie în condiţii de repaus alimentar > 250 mg/dl sau 13,9 mmol/l), trebuie amânată administrarea atezolizumab. Tratamentul poate fi reluat după ce se obţine controlul metabolic cu terapie de substituţie cu insulină.
Meningoencefalită mediată-imun
În studiile clinice, s-au observat cazuri de meningoencefalită în asociere cu atezolizumab (vezi pct. 4.8). Pacienţii trebuie monitorizaţi pentru apariţia semnelor şi simptomelor de meningită sau encefalită.
Tratamentul cu atezolizumab trebuie întrerupt definitiv în cazul meningitei sau encefalitei de orice grad. Trebuie iniţiată corticoterapia administrată intravenos (1-2 mg/kg şi zi metilprednisolon sau echivalent). Imediat ce simptomele se ameliorează, trebuie urmat tratamentul cu prednison în doză de 1-2 mg/kg şi zi sau echivalent.
Neuropatie mediată-imun
La pacienţi trataţi cu atezolizumab, s-au observat sindrom miastenic/miastenie gravis sau sindrom Guillain-Barré, care pot pune viaţa în pericol. Pacienţii trebuie monitorizaţi pentru apariţia semnelor şi simptomelor de neuropatie motorie sau senzitivă.
Tratamentul cu atezolizumab trebuie întrerupt definitiv în cazul sindromului miastenic/miasteniei gravis sau sindromului Guillain-Barré de orice grad. Trebuie luată în considerare iniţierea corticoterapiei sistemice (cu prednison în doză de 1-2 mg/kg şi zi sau echivalent).
Pancreatită mediată-imun
În studiile clinice, s-a observat pancreatită, incluzând creşterea valorilor serice ale amilazei şi lipazei, în asociere cu atezolizumab (vezi pct. 4.8). Pacienţii trebuie monitorizaţi atent pentru apariţia semnelor şi simptomelor sugestive de pancreatită acută.
Tratamentul cu atezolizumab trebuie amânat în cazul valorilor serice crescute de grad ≥ 3 ale amilazei sau lipazei (> 2 x LSVN) sau pancreatitei de grad 2 sau 3 şi trebuie iniţiată corticoterapia administrată intravenos (1-2 mg/kg şi zi metilprednisolon sau echivalent). Imediat ce simptomele se ameliorează, trebuie instituit tratamentul cu prednison în doză de 1-2 mg/kg şi zi sau echivalent. Tratamentul cu atezolizumab poate fi reluat atunci când valorile serice ale amilazei şi lipazei se ameliorează până la grad ≤ 1 într-un interval de 12 săptămâni sau simptomele de pancreatită se remit şi corticoterapia a fost redusă până la ≤ 10 mg pe zi prednison sau echivalent. Tratamentul cu atezolizumab trebuie întrerupt
definitiv în cazul pancreatitei de grad 4 sau al pancreatitei recurente de orice grad.
Reacţii asociate perfuziei
Reacţiile asociate perfuziei au fost observate în studiile clinice cu atezolizumab (vezi pct. 4.8). Viteza de perfuzare trebuie redusă sau tratamentul trebuie întrerupt la pacienţii cu reacţii de grad 1 sau 2. Tratamentul cu atezolizumab trebuie întrerupt definitiv la pacienţii cu reacţii asociate perfuziei de grad 3 sau 4. Pacienţii cu reacţii asociate perfuziei de grad 1 sau 2 pot continua tratamentul cu atezolizumab sub monitorizare atentă; poate fi luată în considerare premedicaţia cu antipiretic şi antihistaminice.
Pacienţi excluşi din studiile clinice
Au fost excluşi din studiile clinice pacienţii cu următoarele afecţiuni: boală autoimună în antecedente,pneumonită în antecedente, metastaze cerebrale active, infecţie cu HIV, hepatită B sau hepatită C.
Pacienţii cărora li s-a administrat un vaccin cu virus viu atenuat în ultimele 28 zile înainte de înrolare, medicamente imunostimulatoare pe cale sistemică în ultimele 4 săptămâni sau medicamente imunosupresoare pe cale sistemică în ultimele 2 săptămâni înainte de înrolarea în studiu au fost excluşi din studiile clinice.
Au fost excluşi pacienţii cu status de performanţă ≥ 2 la momentul iniţial (cu excepţia cohortei 1 a studiului GO29293 [IMvigor210] în care au fost înrolaţi pacienţi cu carcinom urotelial care nu erau eligibili pentru tratamentul cu cisplatină şi în care a fost permis un status de performanţă ≥ 2 la momentul iniţial) (vezi pct. 5.1).
În absenţa datelor, atezolizumab trebuie utilizat cu precauţie la aceste categorii de pacienţi după evaluarea atentă a raportului beneficiu-risc pentru fiecare pacient.
Utilizarea atezolizumab în carcinomul urotelial la pacienţii netrataţi anterior care nu sunt consideraţi eligibili pentru tratamentul cu cisplatină
Caracteristicile iniţiale şi de prognostic ale bolii la populaţia din cohorta 1 a studiului IMvigor210 au fost, în general, comparabile cu cele ale pacienţilor din clinică consideraţi că nu ar fi eligibili pentru tratamentul cu cisplatină, dar ar fi eligibili pentru chimioterapia asociată pe bază de carboplatină.
Există date insuficiente pentru subgrupa de pacienţi care nu ar fi eligibili pentru chimioterapie; prin urmare, atezolizumab trebuie utilizat cu precauţie la aceşti pacienţi, după evaluare individuală atentă a raportului beneficiu potenţial-risc.
Cardul de avertizare pentru pacient
Toţi medicii care prescriu Tecentriq trebuie să fie familiarizaţi cu Informaţiile pentru medic şi recomandările privind conduita terapeutică. Medicul prescriptor trebuie să discute cu pacientul riscurile terapiei cu Tecentriq. Pacientului i se va înmâna Cardul de avertizare pentru pacient şi i se va recomanda să îl aibă întotdeauna asupra sa.
4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
Nu s-au efectuat studii dedicate cu atezolizumab privind interacţiunile farmacocinetice. Deoarece atezolizumab este eliminat din circulaţie prin catabolism, nu sunt de aşteptat interacţiuni medicamentoase metabolice.
Trebuie evitată utilizarea de corticosteroizi sistemici sau imunosupresoare sistemice înainte de începerea tratamentului cu atezolizumab din cauza potenţialului de interferenţă cu activitatea farmacodinamică şi eficacitatea atezolizumab. Cu toate acestea, se pot utiliza corticosteroizii sistemici sau alte medicamente imunosupresoare pentru tratamentul reacţiilor adverse mediate imun după începerea tratamentului cu atezolizumab (vezi pct. 4.4).
4.6 Fertilitatea, sarcina şi alăptarea
Femei aflate la vârsta fertilă
Femeile aflate la vârsta fertilă trebuie să utilizeze măsuri contraceptive eficace în timpul şi timp de 5 luni după tratamentul cu atezolizumab.
Sarcina
Nu există date provenite din utilizarea atezolizumab la femeile gravide. Nu s-au efectuat studii cu atezolizumab privind dezvoltarea şi funcţia de reproducere. Studiile la animale efectuate la modele murine de sarcină au evidenţiat că inhibarea căii PD-L1/PD-1 poate duce la respingerea mediată imun a fetusului în dezvoltare, provocând deces fetal (vezi pct. 5.3). Aceste rezultate indică un risc potenţial, pe baza mecanismului de acţiune, sugerând că administrarea atezolizumab în timpul sarcinii poate avea efecte dăunătoare asupra fătului, incluzând rate crescute de avort şi naştere de feţi decedaţi.
Este cunoscut faptul că imunoglobulinele umane G1 (IgG1) traversează placenta, iar atezolizumab este o IgG1; prin urmare, este posibil ca atezolizumab să se transmită de la mamă la fătul în dezvoltare.
Atezolizumab nu trebuie utilizat în timpul sarcinii, cu excepţia cazului în care starea clinică a femeii impune tratament cu atezolizumab.
Alăptarea
Nu se cunoaşte dacă atezolizumab se excretă în laptele uman. Atezolizumab este un anticorp monoclonal şi este de aşteptat să fie prezent în primul lapte şi ulterior în cantităţi mai mici. Nu se poate exclude un risc pentru nou-născuţi/sugari. Trebuie luată decizia fie de a întrerupe alăptarea, fie de a întrerupe tratamentul cu Tecentriq, având în vedere beneficiul alăptării pentru copil şi beneficiul tratamentului pentru femeie.
Fertilitatea
Nu sunt disponibile date privind efectele posibile ale atezolizumab asupra fertilităţii. Nu s-au efectuat studii cu atezolizumab privind efecte toxice asupra dezvoltării şi funcţiei de reproducere; cu toate acestea, pe baza unui studiu cu durata de 26 săptămâni privind toxicitatea după doze repetate, atezolizumab a avut un efect asupra ciclului menstrual, la o ASC estimată de aproximativ 6 ori ASC la pacienţii trataţi cu doza recomandată şi acest efect a fost reversibil (vezi pct. 5.3). Nu au existat efecte asupra organelor de reproducere masculine.
4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
Tecentriq are o influenţă mică asupra capacităţii de a conduce vehicule sau de a folosi utilaje. Pacienţii care prezintă fatigabilitate trebuie sfătuiţi să nu conducă vehicule şi să nu folosească utilaje până la dispariţia simptomelor (vezi pct. 4.8).
4.8 Reacţii adverse
Rezumatul profilului de siguranţă
Siguranţa Tecentriq se bazează pe datele cumulate de la 2160 pacienţi cu CU metastazat şi NSCLC
metastazat. Cele mai frecvente reacţii adverse au fost fatigabilitate (35,4%), scădere a apetitului
alimentar (25,5%), greaţă (22,9%), dispnee (21,8%), diaree (18,6%), erupţii cutanate tranzitorii
(18,6%), febră (18,3%), vărsături (15,0%), artralgie (14,2%), astenie (13,8%) şi prurit (11,3%).
Lista sub formă de tabel a reacţiilor adverse
Reacţiile adverse (RA) sunt enumerate mai jos în funcţie de clasificarea MedDRA pe aparate, sisteme
şi organe (ASO) şi în funcţie de frecvenţă. Au fost utilizate următoarele categorii de frecvenţe: foarte
frecvente (≥1/10); frecvente (≥1/100 și <1/10); mai puţin frecvente (≥1/1000 şi <1/100); rare
(≥1/10000 şi <1/1000); foarte rare (< 1/10000). În cadrul fiecărei grupe de frecvenţă, reacţiile adverse
sunt prezentate în ordinea descrescătoare a gravităţii.
Tabelul 2: Rezumatul reacţiilor adverse apărute la pacienţi trataţi cu Tecentriq în studiile
clinice
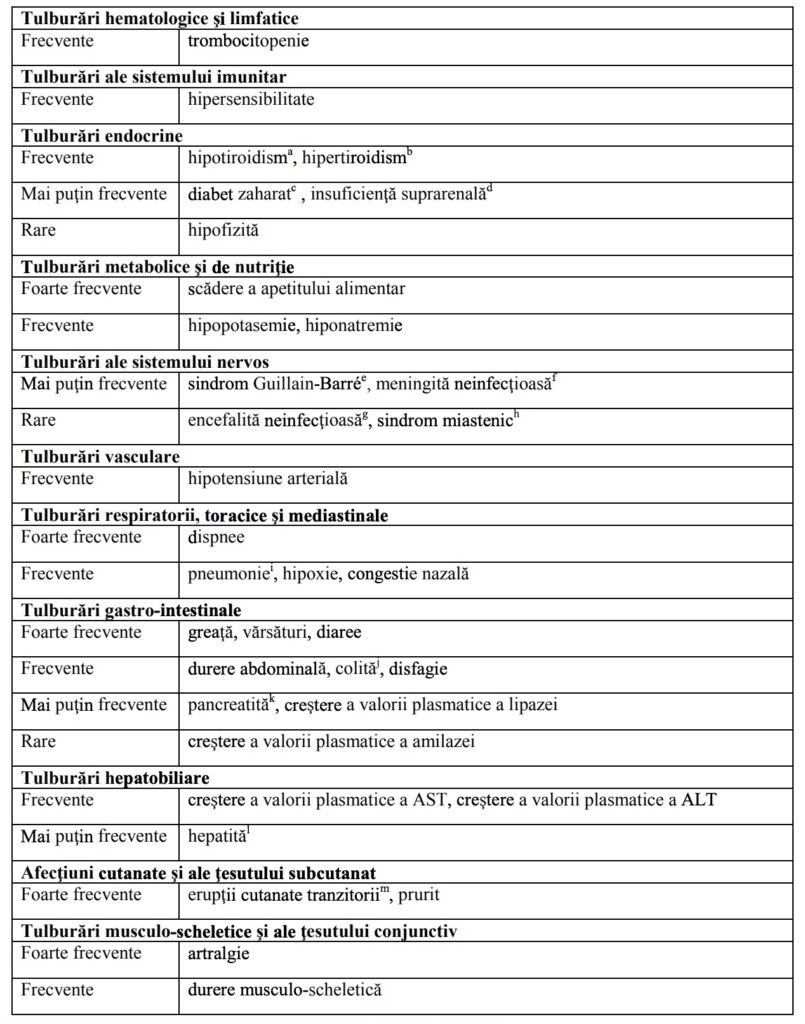

a Include raportările de hipotiroidism, valori serice crescute ale hormonului de stimulare tiroidiană, tiroidită, valori serice scăzute ale hormonului de stimulare tiroidiană, mixedem, valori anormale ale testelor funcţiei tiroidiene, tiroidită acută, valori scăzute ale tiroxinei.
b Include raportările de hipertiroidism, valori serice crescute ale hormonului de stimulare tiroidiană, tiroidită, valori serice scăzute ale hormonului de stimulare tiroidiană, oftalmopatie endocrină, exoftalmie, valori anormale ale testelor funcţiei tiroidiene, tiroidită acută, valori scăzute ale tiroxinei.
c Include raportările de diabet zaharat şi diabet zaharat tip 1.
d Include raportările de insuficienţă suprarenală, insuficienţă suprarenală primară şi boală Addison.
e Include raportările de sindrom Guillain-Barré şi polineuropatie demielinizantă.
f Include raportările de meningită.
g Include raportările de encefalită. h Raportate în studii clinice, altele decât cele la pacienţi cu CU metastazat şi NSCLC metastazat. Frecvenţa se bazează pe expunerea a 6000 pacienţi în toate studiile clinice cu atezolizumab.
i Include raportările de pneumonită, infiltrare pulmonară, bronşiolită, boală pulmonară interstiţială, pneumonită
de iradiere.
j Include raportările de colită, colită autoimună, colită ischemică, colită microscopică.
k Include raportările de pancreatită şi pancreatită acută.
l Include raportările de hepatită autoimună, hepatită, hepatită acută.
m Include raportările de acnee, eczemă, eritem, eritem al pleoapei, eritem polimorf, erupţii cutanate exfoliative, erupţii cutanate ale pleoapei, foliculită, furunculoză, dermatită, dermatită acneiformă, dermatită alergică, dermatită buloasă, dermatită exfoliativă, erupţie indusă de medicament, sindrom de eritrodisestezie palmoplantară, erupţii cutanate tranzitorii, erupţii cutanate eritematoase, erupţii cutanate generalizate, erupţii cutanate maculare, erupţii cutanate maculo-papuloase, erupţii cutanate papuloase, erupţii cutanate papulo-scuamoase, erupţii cutanate pruriginoase, erupţii cutanate pus
Descrierea reacţiilor adverse selectate
Datele de mai jos reflectă expunerea la atezolizumab în studiile clinice pentru reacţiile adverse semnificative clinic (vezi pct. 5.1). Recomandările privind conduita terapeutică pentru aceste reacţii adverse sunt descrise la pct. 4.2 şi 4.4.
Pneumonită mediată-imun
Pneumonita a survenit la 3,1% (68/2160) dintre pacienţii trataţi cu atezolizumab pentru CU metastazat şi NSCLC metastazat. Dintre cei 68 pacienţi, unul a prezentat un eveniment letal. Timpul median până la debut a fost de 3,5 luni (interval 3 zile până la 20,5 luni). Durata mediană a fost de 1,5 luni (interval 0 zile până la 15,1+ luni; + denotă o valoare cenzurată). Pneumonita a dus la oprirea tratamentului cu atezolizumab la 10 (0,5%) pacienţi. Pneumonita care a necesitat administrarea de corticoterapie a survenit la 1,6% (34/2160) dintre pacienţii trataţi cu atezolizumab.
Hepatită mediată-imun
Hepatita a survenit la 0,3% (7/2160) dintre pacienţii trataţi cu atezolizumab pentru CU metastazat şi NSCLC metastazat. Timpul median până la debut a fost de 1,1 luni (interval 9 zile până la 7,9 luni). Durata mediană a fost de 1 lună (interval 9 zile până la 1,9+ luni; + denotă o valoare cenzurată). Hepatita a dus la oprirea tratamentului cu atezolizumab la 2 (< 0,1%) pacienţi. Hepatita care a necesitat administrarea de corticoterapie a survenit la 0,2% (5/2160) dintre pacienţii trataţi cu atezolizumab.
Colită mediată-imun
Colita a survenit la 1,1% (23/2160) dintre pacienţii trataţi cu atezolizumab pentru CU metastazat şi NSCLC metastazat. Timpul median până la debut a fost de 4 luni (interval 15 zile până la 15,2 luni). Durata mediană a fost de 1,4 luni (interval 3 zile până la 17,8+ luni; + denotă o valoare cenzurată). Colita a dus la oprirea tratamentului cu atezolizumab la 5 (0,2%) pacienţi. Colita care a necesitat administrarea de corticoterapie a survenit la 0,5% (10/2160) dintre pacienţii trataţi cu atezolizumab.
Endocrinopatii mediate-imun
Hipotiroidismul a survenit la 4,7% (101/2160) dintre pacienţii trataţi cu atezolizumab pentru CU metastazat şi NSCLC metastazat. Timpul median până la debut a fost de 5,5 luni (interval 15 zile până la 31,3 luni). Hipertiroidismul a survenit la 1,7% (36/2160) dintre pacienţii trataţi cu atezolizumab pentru CU metastazat și NSCLC. Timpul median până la debut a fost de 3,5 luni (interval 21 zile până la 31,3 luni). Insuficienţa suprarenală a survenit la 0,3% (7/2160) dintre pacienţii trataţi cu atezolizumab pentru CU
metastazat și NSCLC metastazat. Timpul median până la debut a fost de 5,7 luni (interval: 3 zile până la 19 luni). Insuficienţa suprarenală care a necesitat administrarea de corticoterapie a survenit la 0,3% (6/2160) dintre pacienţii trataţi cu atezolizumab.
Hipofizita a survenit la < 0,1% (1/2160) dintre pacienţii trataţi cu atezolizumab pentru CU metastazat şi NSCLC metastazat. Timpul până la debut pentru acest pacient a fost de 13,7 luni. Diabetul zaharat a survenit la 0,3% (6/2160) dintre pacienţii trataţi cu atezolizumab pentru CU metastazat și NSCLC metastazat. Timpul median până la debut a fost de 3 zile până la 6,5 luni. Diabetul zaharat a dus la oprirea tratamentului cu atezolizumab la 1 (<0,1%) pacient.
Meningoencefalită mediată-imun
Meningita a survenit la 0,1% (3/2160) dintre pacienţii trataţi cu atezolizumab pentru CU metastazat şi NSCLC metastazat. Timpul median până la debut a fost de 15 până la 16 zile. La toţi cei trei pacienţi a fost necesară administrarea de corticoterapie şi oprirea tratamentului cu atezolizumab.
Encefalita a survenit la <0,1% (2/2160) dintre pacienţii care au primit atezolizumab pentru CU metastazat şi NSCLC metastazat. Timpul până la debut a fost de 14 şi 16 zile. Encefalita a dus la oprirea tratamentului cu atezolizumab la 1 (< 0,1%) pacient. Encefalita care a necesitat administrarea de corticoterapie a survenit la < 0,1% (1/2160) dintre pacienţii trataţi cu atezolizumab.
Neuropatie mediată-imun
Sindromul Guillain-Barré şi polineuropatia demielinizantă au survenit la 0,2% (5/2160) dintre pacienţii tratați cu atezolizumab pentru CU metastazat şi NSCLC metastazat. Timpul median până la debut a fost de 7 luni (interval: 18 zile până la 8,1 luni). Durata mediană a fost de 4,6 luni (0+ zile până la 8,3+ luni; + denotă o valoare cenzurată). Sindromul Guillain- Barré a dus la oprirea tratamentului cu atezolizumab la 1 pacient (<0,1%). Sindromul Guillain- Barré care a necesitat administrarea de corticoterapie a survenit la <0,1% (2/2160) dintre pacienţii tratați cu atezolizumab.
Sindrom miastenic
Miastenia gravis a survenit la < 0,1% (4/6000) dintre pacienţii trataţi cu atezolizumab în toate studiile clinice pentru multiple tipuri de tumori. Timpul până la debut a variat între 20 zile şi 4 luni. Tratamentul cu atezolizumab a fost oprit la toţi cei patru pacienţi. Sindromul miastenic/miastenia gravis care a necesitat administrarea de corticoterapie a survenit la < 0,1% (3/6000) dintre pacienţii trataţi cu atezolizumab.
Pancreatită mediată-imun
Pancreatita, incluzând creşterea valorilor serice ale amilazei şi lipazei, a survenit la 0,5% (10/2160) dintre pacienţii trataţi cu atezolizumab pentru CU metastazat şi NSCLC metastazat. Timpul median până la debut a fost de 5,5 luni (interval: 9 zile până la 16,9 luni). Durata mediană a fost de 19 zile (interval: 3 zile până la 11,2+ luni; + denotă o valoare cenzurată). Pancreatita care a necesitat administrarea de corticoterapie a survenit la <0,1% (2/2160) dintre pacienţii trataţi cu atezolizumab.
Imunogenitate
În studiul IMvigor210, 43,9% dintre pacienţi au avut rezultate pozitive la testarea pentru anticorpi anti-atezolizumab (AAT) la unul sau mai multe momente de evaluare după administrarea dozei. În studiul OAK (GO28915), rata de apariţie a AAT în urma administrării tratamentului a fost 30,4%. În general, pozitivitatea pentru AAT a părut să nu aibă un impact semnificativ clinic asupra farmacocineticii, eficacităţii şi siguranţei.
Nu sunt disponibile date pentru a permite formularea unor concluzii referitoare la orice posibil efect al anticorpilor neutralizanţi.
Raportarea reacţiilor adverse suspectate
Este importantă raportarea reacţiilor adverse suspectate după autorizarea medicamentului. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniştii din domeniul sănătăţii sunt rugaţi să raporteze orice reacţie adversă suspectată prin intermediul sistemului naţional de raportare, astfel cum este menţionat în Anexa V.
5. PROPRIETĂŢI FARMACOLOGICE
5.1 Proprietăţi farmacodinamice
Grupa farmacoterapeutică: Agenţi antineoplazici, anticorpi monoclonali, codul ATC: încă nealocat
Mecanism de acţiune
Ligandul 1 cu rol în controlul morţii celulare programate (PD-L1) poate fi exprimat pe suprafaţa celulelor tumorale şi/sau celulelor imune care infiltrează tumora şi poate contribui la inhibarea răspunsului imunitar antitumoral în micromediul tumoral. Legarea PD-L1 de receptorii PD-1 şi B7.1 prezenţi pe suprafaţa limfocitelor T şi celulelor prezentatoare de antigen suprimă activitatea citotoxică a limfocitelor T, proliferarea limfocitelor T şi producţia de citokine.
Atezolizumab este un anticorp monoclonal umanizat de tip imunoglobulină G1 (IgG1) modificat la nivelul regiunii Fc, care se leagă direct de PD-L1 şi creează o blocadă dublă a receptorilor PD-1 şi B7.1, contracarând astfel inhibarea mediată pe calea PD-L1/PD-1 a răspunsului imunitar şi reactivând răspunsul imun antitumoral, fără a induce citotoxicitate celulară dependentă de anticorpi. Atezolizumab nu influenţează interacţiunea PD-L2/PD-1, permiţând semnalelor inhibitorii mediate pe calea PD-L2/PD-1 să persiste.
Eficacitate clinică şi siguranţă
Durata tratamentului
În cazul pacienţilor care nu au fost trataţi anterior, tratamentul cu Tecentriq a fost permis până la progresia bolii.
În cazul pacienţilor care au fost trataţi anterior în studiile clinice pivot, tratamentul cu Tecentriq a fost permis până la pierderea beneficiului clinic, aşa cum a fost definit acesta, pe baza următoarelor criterii:
- Absenţa simptomelor şi semnelor (incluzând agravarea valorilor parametrilor de laborator [de exemplu, hipercalcemie nou apărută sau agravată]) indicând progresia inechivocă a bolii
- Nicio scădere a scorului de performanţă ECOG
- Absenţa progresiei tumorale în localizările anatomice critice (de exemplu, boală
leptomeningeală) care nu pot fi abordate cu uşurinţă şi stabilizate prin intervenţii medicale permise prin protocol înainte de administrarea repetată a dozelor - Dovada beneficiului clinic, evaluat de către investigator
Carcinomul urotelial
IMvigor211 (GO29293): Studiu clinic în CU local avansat sau metastazat la pacienţi adulţi după tratament anterior cu chimioterapie
Un studiu clinic de fază III, deschis, multicentric, internaţional, randomizat (IMvigor211) a fost efectuat pentru a evalua eficacitatea şi siguranţa atezolizumab, comparativ cu chimioterapia (vinflunină, docetaxel sau paclitaxel la alegerea investigatorului) la pacienţi cu CU local avansat sau metastazat, care au avut progresie pe durata tratamentului sau după o cură de tratament pe bază de săruri de platină. Din acest studiu au fost excluşi pacienţii care aveau antecedente de boală autoimună; metastaze cerebrale active sau dependente de corticoterapie; administrare de vaccin cu virus viu atenuat în ultimele 28 zile înainte de înrolare; şi administrare sistemică de medicamente imunostimulatoare într-un interval de 4 săptămâni sau administrare sistemică de medicamente imunosupresoare într-un interval de 2 săptămâni înainte de înrolare. Evaluările tumorale au fost efectuate la fiecare 9 săptămâni în primele 54 săptămâni şi la fiecare 12 săptămâni după aceea. Au fost evaluate prospectiv specimene de ţesut tumoral pentru a testa expresia PD-L1 la nivelul celulelor imune care infiltrează tumora (CI), iar rezultatele au fost utilizate pentru a defini subgrupurile în funcţie de expresia PD-L1 pentru analizele descrise mai jos.
În total, au fost înrolaţi 931 pacienţi. Pacienţii au fost randomizaţi (1:1) pentru a li se administra fie atezolizumab, fie chimioterapie. Randomizarea a fost stratificată în funcţie de chimioterapie (vinflunină versus taxan), statusul expresiei PD-L1 la nivelul CI (< 5% versus ≥ 5%), numărul factorilor prognostici de risc (0 versus 1-3) şi prezenţa metastazelor hepatice (da versus nu). Factorii prognostici de risc au inclus timpul < 3 luni de la administrarea anterioară a chimioterapiei, statusul de performanţă ECOG > 0 şi concentraţia hemoglobinei< 10 g/dl.
Atezolizumab a fost administrat în doză fixă de 1200 mg prin perfuzie intravenoasă la interval de 3 săptămâni. Nu a fost permisă reducerea dozei. Pacienţii au fost trataţi până la pierderea beneficiului clinic, aşa cum a fost evaluat de către investigator sau până la apariţia toxicităţii inacceptabile. Vinflunina a fost administrată în doză de 320 mg/m2 prin perfuzie intravenoasă în ziua 1 a fiecărui ciclu de 3 săptămâni până la progresia bolii sau apariţia toxicităţii inacceptabile. Paclitaxel a fost administrat în doză de 175 mg/m2 prin perfuzie intravenoasă cu durata de 3 ore în ziua 1 a fiecărui ciclu de 3 săptămâni până la progresia bolii sau apariţia toxicităţii inacceptabile. Docetaxel a fost administrat în doză de 75 mg/m2 prin perfuzie intravenoasă în ziua 1 a fiecărui ciclu de 3 săptămâni până la progresia bolii sau apariţia toxicităţii inacceptabile. Pentru toţi pacienţii trataţi, durata mediană a tratamentului a fost de 2,8 luni pentru pacienţii din braţul tratat cu atezolizumab, de 2,1 luni pentru pacienţii din braţele tratate cu vinflunină şi paclitaxel şi de 1,6 luni pentru cei din braţul tratat cu docetaxel.
Caracteristicile demografice şi caracteristicile bolii la momentul iniţial al analizei populaţionale primare au fost bine echilibrate între braţele de tratament. Vârsta mediană a fost de 67 ani (interval: 31 până la 88) şi 77,1% dintre pacienţi au fost bărbaţi. Majoritatea pacienţilor au fost caucazieni (72,1%), 53,9% dintre pacienţii din braţul tratat cu chimioterapie au fost trataţi cu vinflunină, 71,4% dintre pacienţi au avut cel puţin un factor de risc de prognostic nefavorabil şi 28,8% au avut metastaze hepatice la momentul iniţial. Scorul de performanţă ECOG la momentul iniţial a fost 0 (45,6%) sau 1 (54,4%). Vezica urinară a fost localizarea pentru tumora primară la 71,1% dintre pacienţi şi 25,4% dintre pacienţi au avut carcinom al tractului urinar superior. O proporţie de 24,2%
dintre pacienţi care primiseră anterior numai terapie adjuvantă sau neoadjuvantă pe bază de săruri de platină au avut progresie a bolii într-un interval de 12 luni.
Criteriul principal de evaluare a eficacităţii în cadrul studiului IMvigor211 a fost supravieţuirea generală (SG). Criteriile secundare de evaluare a eficacităţii, aşa cum a fost evaluată de către investigator, utilizând Criteriile de Evaluare a Răspunsului în Tumorile Solide (RECIST, Response Evaluation Criteria in Solid Tumors) versiunea 1.1, au fost rata de răspuns obiectiv (RRO), supravieţuirea fără progresia bolii (SFP) şi durata răspunsului (DR). Comparaţiile în ceea ce priveşte SG între braţul de tratament şi cel de control în cadrul populaţiilor CI2/3, CI1/2/3 şi ITT (în intenţie de tratament, de exemplu toţi pacienţii eligibili netestaţi) au fost evaluate utilizând o procedură ierarhică cu secvenţă fixă bazată pe un test stratificat log rank la un nivel de semnificaţie bilateral de 5% după cum urmează: pasul 1) populaţia CI2/3; pasul 2) populaţia CI1/2/3; pasul 3) populaţia alcătuită din toţi
pacienţii eligibili netestaţi. Rezultatele privind SG pentru fiecare dintre paşii 2 şi 3 au putut fi evaluate oficial pentru semnificaţia statistică numai dacă rezultatul obţinut la pasul anterior a fost semnificativ statistic.
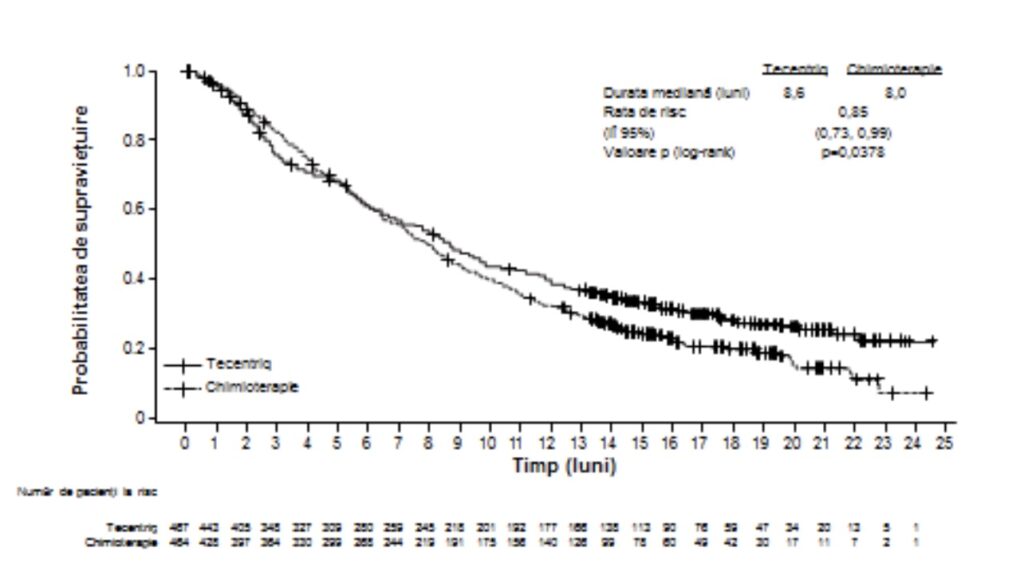
Durata mediană a urmăririi privind supravieţuirea a fost de 17 luni. Analiza primară a studiului IMvigor211 nu şi-a atins obiectivul principal privind SG. Atezolizumab nu a demonstrat un beneficiu semnificativ statistic privind supravieţuirea, comparativ cu chimioterapia la pacienţii cu carcinom urotelial local avansat sau metastazat trataţi anterior. În ceea ce priveşte ordinea evaluării ierarhice specificată anterior, populaţia CI2/3 a fost testată prima, cu o valoare a RR în ceea ce priveşte SG de 0,87 (IÎ 95%: 0,63; 1,21; SG mediană de 11,1 luni comparativ cu 10,6 luni pentru atezolizumab şi, respectiv chimioterapie). Valoarea p stratificată log rank a fost de 0,41 şi, prin urmare, rezultatele sunt considerate fără semnificaţie statistică la această populaţie de pacienţi. În consecinţă, nu au putut fi efectuate teste dedicate privind semnificaţia statistică pentru SG la nivelul populaţiei CI1/2/3 sau la nivelul populaţiei alcătuite din toţi pacienţii eligibili netestaţi, iar rezultatele acestor analize sunt considerate exploratorii. Rezultatele cheie în populaţia alcătuită din toţi pacienţii eligibili netestaţi sunt rezumate în tabelul 3. Curba Kaplan-Meier pentru SG în populaţia alcătuită din toţi pacienţii eligibili netestaţi este prezentată în Figura 1.
Tabelul 3: Rezumatul datelor privind eficacitatea la toţi pacienţii eligibili netestaţi din cadrul studiului IMvigor211
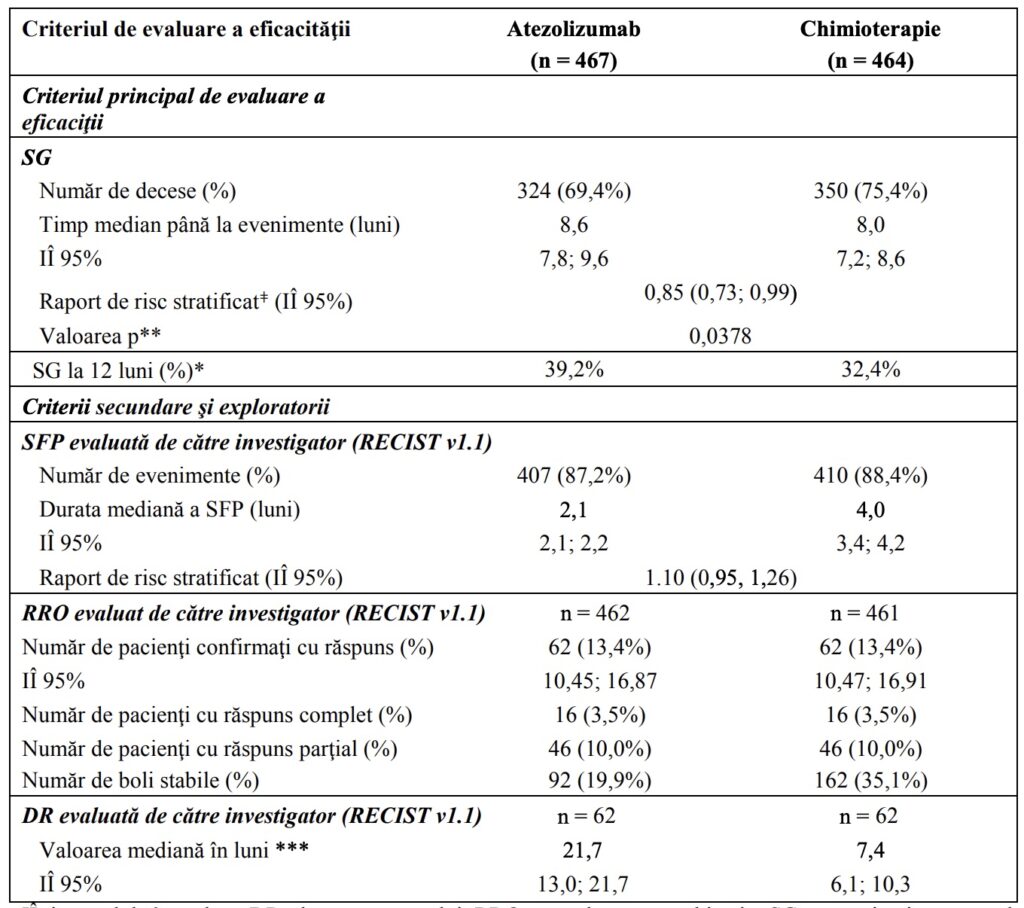
IÎ=interval de încredere; DR=durata răspunsului; RRO= rata de răspuns obiectiv; SG=supravieţuirea generală; SFP=supravieţuirea fără progresie; RECIST=Criteriile pentru evaluarea răspunsului în tumorile solide v1.1.
Pe baza estimării estimării Kaplan-Meier
ǂ Stratificaţi în funcţie de chimioterapie (vinflunină versus taxan), statusul privind CI (<5% versus ≥ 5%), numărul factorilor prognostici de risc (0 versus 1-3) şi prezenţa metastazelor hepatice (da versus nu).
** Pe baza testului stratificat log-rank; furnizat numai în scop descriptiv; conform analizei ierarhice specificată anterior, valoarea p pentru analiza SG la populaţia alcătuită din toţi pacienţii eligibili netestaţi nu poate fi considerată semnificativă statistic.
*** Răspunsurile erau prezente la 63% dintre respondenţi în grupul de tratament cu atezolizumab şi la 21% dintre respondenţi în grupul cu chimioterapie.
Figura 1: Curba Kaplan-Meier privind supravieţuirea generală (IMvigor211)
IMvigor210 (GO29293): Studiu clinic cu un singur braţ efectuat la pacienţi cu carcinom urotelial netrataţi anterior care nu sunt eligibili pentru terapia cu cisplatină şi la pacienţi cu carcinom urotelial căror li s-a administrat anterior chimioterapie.
Un studiu clinic de fază II, multicentric, internaţional, cu două cohorte, cu un singur braţ, IMvigor210 a fost efectuat la pacienţi cu diagnostic de CU local avansat sau metastazat (cunoscut, de asemenea, sub denumirea de cancer vezical urotelial).
În total, au fost înrolaţi în studiu 438 pacienţi şi au fost două cohorte de pacienţi. Cohorta 1 a inclus pacienţi cu diagnostic de CU local avansat sau metastazat netrataţi anterior care nu erau eligibili pentru, sau nu puteau utiliza chimioterapie pe bază de cisplatină sau care au avut progresie a bolii la cel puţin 12 luni după tratament cu o schemă chimioterapică adjuvantă sau neoadjuvantă pe bază de săruri de platină. Cohorta 2 a inclus pacienţi cărora li s-a administrat cel puţin o schemă chimioterapică pe bază de săruri de platină pentru CU local avansat sau metastazat sau care au avut progresie a bolii pe parcursul a 12 luni de tratament cu o schemă chimioterapică adjuvantă sau neoadjuvantă pe bază de săruri de platină.
În cohorta 1, au fost trataţi 119 pacienţi cu atezolizumab 1200 mg administrat în perfuzie intravenoasă la interval de 3 săptămâni până la progresi bolii. Vârsta mediană a fost de 73 ani. Cei mai mulţi dintre pacienţi au fost bărbaţi (81%) şi majoritatea acestora au fost caucazieni (91%).
Cohorta 1 a inclus 45 pacienţi (38%) cu status de performanţă ECOG de 0, 50 pacienţi (42%) cu status de performanţă ECOG de 1 şi 24 pacienţi (20%) cu status de performanţă ECOG de 2, 35 pacienţi (29%) fără factori de risc Bajorin (status de performanţă ECOG ≥ 2 şi metastaze viscerale), 66 pacienţi (56%) cu un factor de risc Bajorin şi 18 pacienţi (15%) cu doi factori de risc Bajorin, 84 pacienţi (71%) cu insuficienţă renală (rata de filtrare glomerulară [RFG] < 60 mL/min) şi 25 pacienţi (21%) cu metastaze hepatice.
Criteriul principal de evaluare a eficacităţii pentru cohorta 1 a fost rata de răspuns obiectiv confirmat (RRO), aşa cum a fost evaluată de către o unitate independentă de analiză (IRF), utilizând criteriile RECIST versiunea 1.1 (RECIST v1.1).
Analiza primară a fost efectuată când toţi pacienţii au avut cel puţin 24 săptămâni de urmărire. Durata mediană a tratamentului a fost de 15,0 săptămâni, iar durata mediană a urmăririi privind supravieţuirea a fost de 8,5 luni la toţi pacienţii eligibili netestaţi. Au fost demonstrate RRO semnificative clinic, conform evaluării de către IRF pe baza RECISTv1.1; cu toate acestea, la comparaţia cu rata de răspuns prespecificată de 10% din grupul de control istoric, semnificaţia statistică pentru obiectivul principal nu a fost atinsă. RRO confirmate conform IRF-RECIST v1.1 au fost de 21,9% (IÎ 95%: 9,3; 40,0) la pacienţii cu expresie PD-L1 ≥ 5%, de 18,8% (IÎ 95%: 10,9; 29,0) la pacienţii cu expresie PD-L1 ≥ 1%
şi de 19,3% (IÎ 95%: 12,7; 27,6) la toţi pacienţii eligibili netestaţi. Durata mediană a răspunsului (DR) nu a fost atinsă în niciunul dintre subgrupurile cu expresie a PD-L1, nici în grupul cu toţi pacienţii eligibili netestaţi. Datele privind SG nu erau mature la un raport evenimente-pacienţi de aproximativ 40%. Valoarea mediană a SG în toate subgrupurile de pacienţi (expresia PD-L1 ≥ 5 % şi ≥ 1 %) şi la toţi pacienţii eligibili netestaţi a fost de 10,6 luni.
A fost efectuată o analiză actualizată cu o durată mediană de urmărire a supravieţuirii de 17,2 luni pentru cohorta 1, care este prezentată rezumativ în Tabelul 4. Mediana DR nu a fost atinsă în niciunul dintre subgrupurile cu expresie PD-L1 şi nici la toţi pacienţii eligibili netestaţi.
Tabelul 4: Rezumatul datelor actualizate privind eficacitatea (cohorta 1 a studiului IMvigor210)
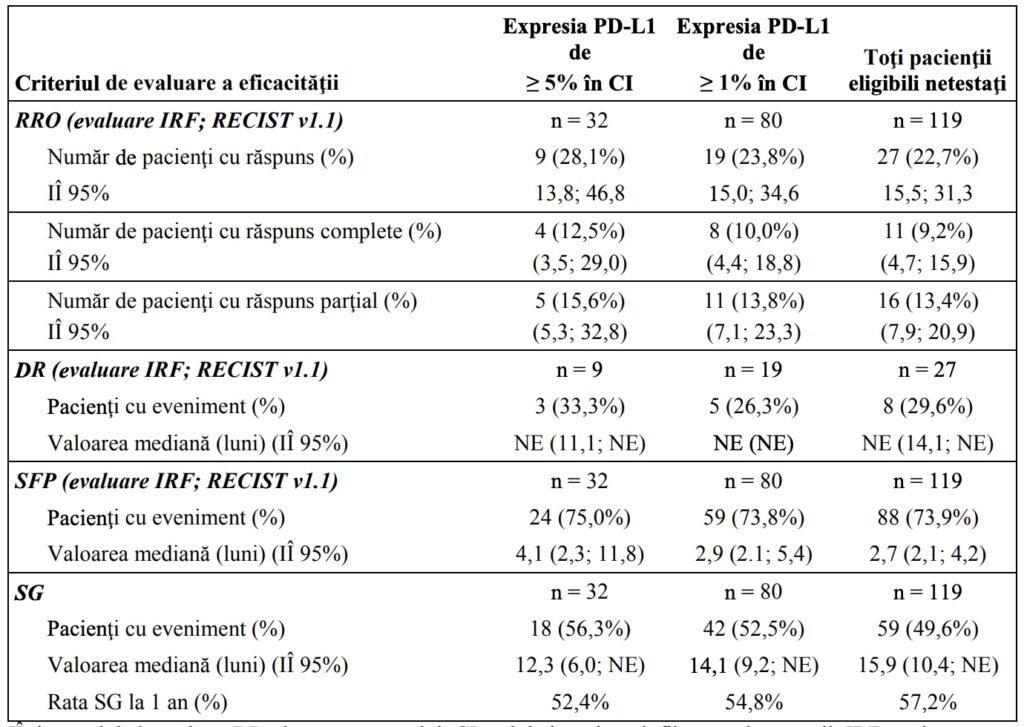
IÎ=interval de încredere; DR=durata răspunsului; CI=celule imunitare infiltrante ale tumorii; IRF= unitate independentă de analiză; NE=nu se poate estima; RRO=rata de răspuns obiectiv; SG=supravieţuirea generală;
SFP=supravieţuirea fără progresie; RECIST= Criteriile pentru evaluarea răspunsului în tumorile solide v1.1.
În cohorta 2, criteriile coprincipale de evaluare a eficacităţii au fost reprezentate de RRO confirmată, aşa cum a fost evaluată de către IRF utilizând criteriile RECIST v.1.1 şi RRO evaluată de către investigator conform criteriilor RECIST modificate (mRECIST). Aceasta a inclus 310 pacienţi trataţi cu atezolizumab 1200 mg administrat în perfuzie intravenoasă la interval de 3 săptămâni, până la pierderea beneficiului clinic. Analiza primară a cohortei 2 a fost efectuată când toţi pacienţii au avut cel puţin 24 săptămâni de urmărire. Studiul a atins obiectivele coprincipale în cohorta 2, demonstrând RRO semnificative statistic, evaluate de IRF conform RECIST v1.1 şi evaluate de investigator conform mRECIST, comparativ cu rata de răspuns prespecificată de 10% din grupul de control istoric.
De asemenea, a fost efectuată o analiză cu o durată mediană de urmărire a supravieţuirii de 21,1 luni pentru cohorta 2. RRO confirmate, aşa cum au fost evaluate de către IRF utilizând criteriile RECIST v.1.1 au fost de 28,0% (IÎ 95%: 19,5; 37,9) la pacienţi cu expresie a PD-L1 ≥ 5%, de 19,3% (IÎ 95%: 14,2; 25,4) la pacienţi cu expresie a PD-L1 ≥ 1% şi de 15,8% (IÎ 95%: 11,9; 20,4) la toţi pacienţii eligibili netestaţi. RRO confirmată prin evaluarea de către investigator, utilizând criteriile mRECIST a fost de 29,0% (IÎ 95%: 20,4; 38,9) la pacienţi cu expresie a PD-L1 ≥ 5%, de 23,7% (IÎ 95%: 18,1; 30,1) la pacienţi cu expresie a PD-L1 ≥ 1% şi de 19,7% (IÎ 95%: 15,4; 24,6) la toţi pacienţii eligibili netestaţi. Rata răspunsului complet conform evaluării de către IRF, utilizând criteriile RECIST v.1.1 la toţi pacienţii eligibili netestaţi a fost de 6,1% (IÎ 95%: 3,7; 9,4). Pentru cohorta 2, valoarea mediană a DR nu a fost atinsă în subgrupul cu expresie PD-L1 şi nici la toţi pacienţii eligibili netestaţi; cu toate acestea, a fost atinsă la pacienţi cu expresie a PD-L1 < 1% (13,3 luni; IÎ 95%, 4,2; NE). Rata SG la 12 luni a fost de 37% la toţi pacienţii eligibili netestaţi.
Cancerul bronho-pulmonar altul decât cu celule mici
OAK (GO28915): Studiu clinic de fază III, randomizat, la pacienţi cu NSCLC local avansat sau
metastazat cărora li s-a administrat anterior chimioterapie.
A fost efectuat un studiu clinic de fază III, deschis, multicentric, internaţional, randomizat, OAK, pentru a evalua eficacitatea şi siguranţa atezolizumab, comparativ cu docetaxel, la pacienţi cu NSCLC local avansat sau metastazat, care au avut progresie pe durata tratamentului sau după o schemă terapeutică pe bază de săruri de platină. Au fost excluşi din acest studiu pacienţii cu antecedente de boală autoimună, metastaze cerebrale active sau dependente de corticoterapie, administrare a unui vaccin cu virus viu atenuat în ultimele 28 zile anterior înrolării, administrare de medicamente imunostimulatoare sistemice în ultimele 4 săptămâni înainte de înrolare sau de medicamente
imunosupresoare sistemice în ultimele 2 săptămâni anterior înrolării. Evaluările tumorale au fost efectuate la fiecare 6 săptămâni în primele 36 săptămâni şi la fiecare 9 săptămâni după aceea. Au fost evaluate prospectiv specimene de ţesut tumoral pentru a testa expresia PD-L1 pe celulele tumorale (CT) şi pe celule imunitare infiltrante ale tumorii (CI).
În total, au fost înrolaţi 1225 pacienţi şi în conformitate cu planul de analiză, primii 850 pacienţi randomizaţi au fost incluşi în analiza de eficacitate primară. Randomizarea a fost stratificată în funcţie de statusul expresiei PD-L1 la nivelul CI, de numărul schemelor anterioare de chimioterapie şi de profilul histologic. Pacienţii au fost randomizaţi (1:1) pentru a li se administra atezolizumab sau docetaxel.
Atezolizumab a fost administrat în doză fixă de 1200 mg prin perfuzie intravenoasă la interval de 3 săptămâni. Nu a fost permisă reducerea dozei. Pacienţii au fost trataţi până la pierderea beneficiului clinic, aşa cum a fost evaluat de către investigator. Docetaxel a fost administrat în doză de 75 mg/m2 prin perfuzie intravenoasă în ziua 1 a fiecărui ciclu de 3 săptămâni, până la progresia bolii. Pentru toţi pacienţii trataţi, durata mediană a tratamentului a fost de 2,1 luni pentru pacienţii din braţul tratat cu docetaxel şi de 3,4 luni pentru cei din braţul tratat cu atezolizumab.
Caracteristicile demografice şi caracteristicile bolii la momentul iniţial al analizei populaţionale primare au fost bine echilibrate între braţele de tratament. Vârsta mediană a fost de 64 ani (interval: 33 până la 85) şi 61% dintre pacienţi au fost bărbaţi. Majoritatea pacienţilor au fost caucazieni (70%). Aproximativ trei sferturi dintre pacienţi au avut histologie cu aspect non-scuamos (74%) 10% au avut mutaţie cunoscută EGFR, 0,2% au avut mutaţii conformaţionale ALK cunoscute, 10% au avut metastaze SNC la momentul iniţial şi majoritatea pacienţilor erau fumători sau foşti fumători (82%). Scorul de performanţă ECOG la momentul iniţial a fost 0 (37%) sau 1 (63%). În total, 75% dintre
pacienţi au fost trataţi anterior cu o singură schemă terapeutică pe bază de săruri de platină.
Criteriul principal de evaluare a eficacităţii a fost SG. Rezultatele principale ale acestui studiu cu o durată mediană de urmărire a supravieţuirii de 21 luni sunt prezentate rezumativ în Tabelul 5. Curbele Kaplan-Meier pentru SG în populaţia ITT sunt prezentate în Figura 2. Figura 3 rezumă rezultatele SG în subgrupurile ITT şi PD-L1, demonstrând beneficiul de SG cu atezolizumab în toate subgrupurile, incluzându-le pe cele cu expresie PD-L1 < 1% la nivelul CT şi CI.
Tabelul 5: Rezumatul datelor privind eficacitatea din analiza populaţională primară (toţi
pacienţii eligibili netestaţi)* (OAK)
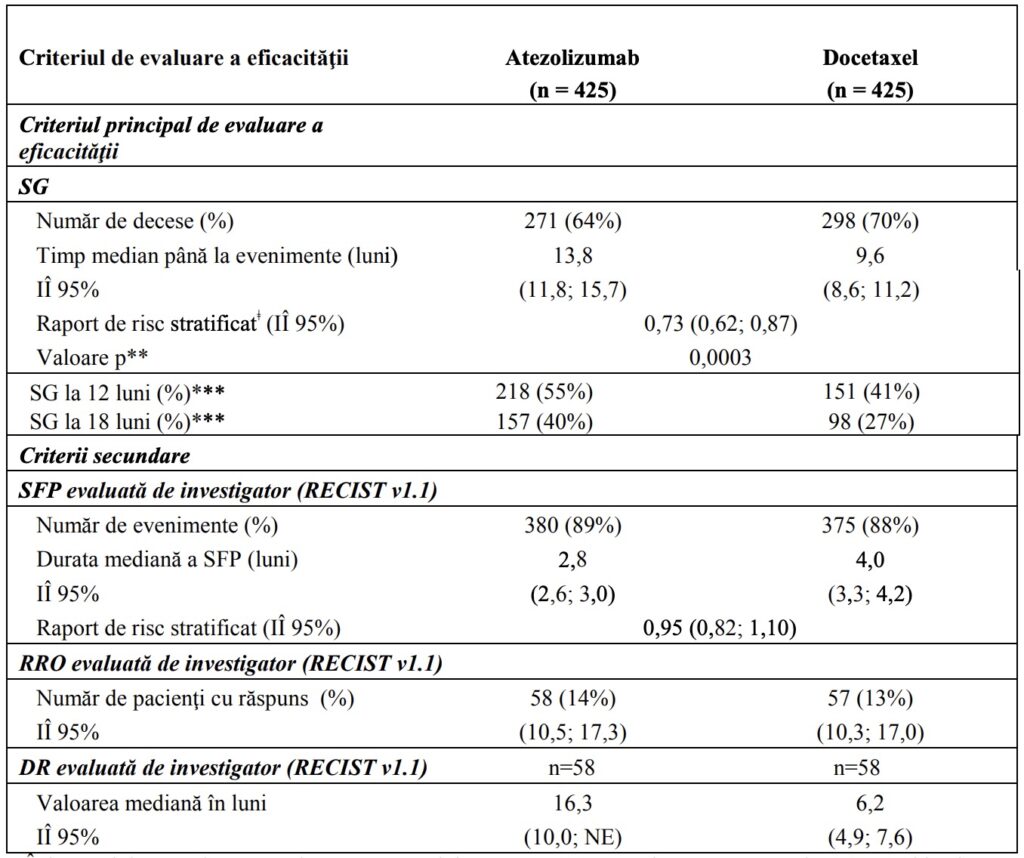
IÎ=interval de încredere; DR=durata răspunsului; NE=nu se poate estima; RRO=rata de răspuns obiectiv; SG=supravieţuirea generală; SFP=supravieţuirea fără progresie; RECIST= Criteriile pentru evaluarea răspunsului în tumorile solide v1.1.
*Populaţia vizată de analiza primară constând din primii 850 pacienţi randomizaţi
ǂStratificat în funcţie de expresia PD-L1 în celule imunitare infiltrante ale tumorii, numărul de regimuri anterioare de chimioterapie şi histologie
** Pe baza testului log-rank stratificat
*** Pe baza estimărilor Kaplan Meier
Figura 2: Curba Kaplan-Meier pentru supravieţuirea generală în populaţia vizată de analiza
primară (toţi subiecţii eligibili netestaţi) (OAK)
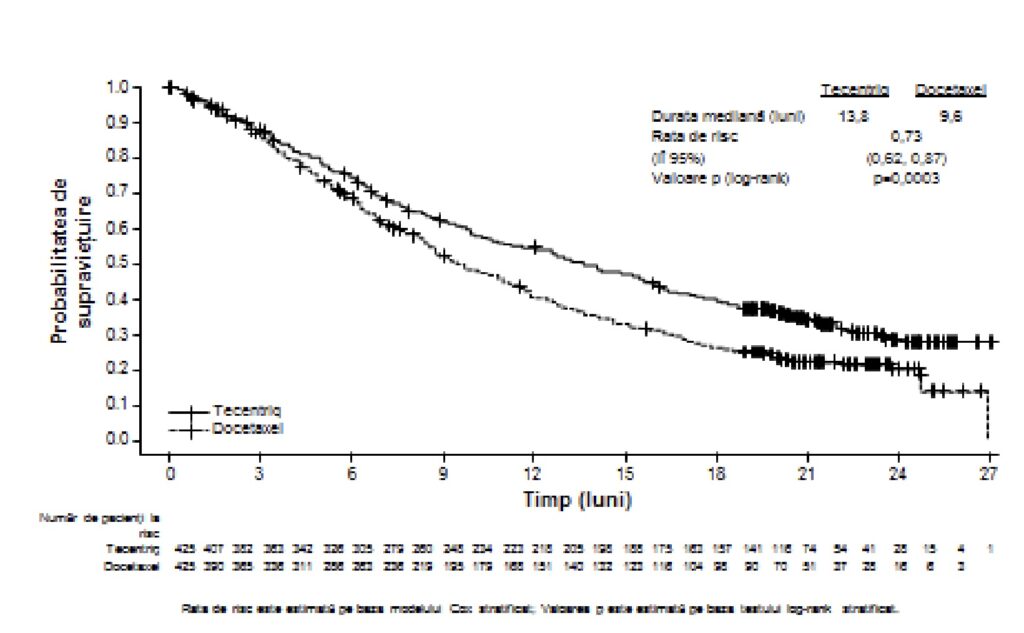
Figura 3: Diagrama supravieţuirii generale în funcţie de expresia PD-L1 la nivelul populaţiei
vizate de analiza primară (OAK)
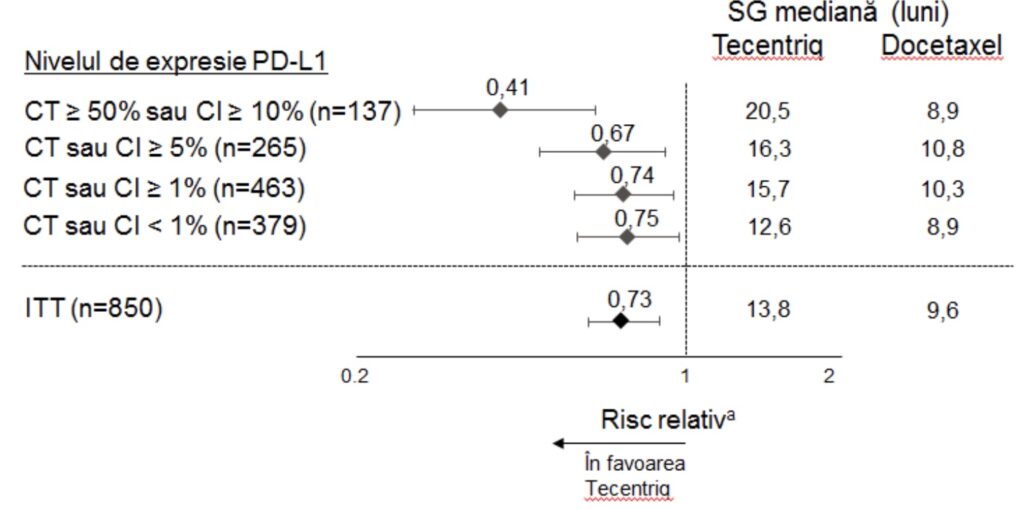
a RR stratificat pentru ITT și CT sau CI ≥ 1%. RR nestratificat pentru alte subgrupuri.
A fost observată o îmbunătăţire a SG cu atezolizumab, comparativ cu docetaxel, atât la pacienţii cu NSCLC non-scuamos (raport de risc [RR] de 0,73, IÎ 95%: 0,60; 0,89; SG mediană de 15,6 comparativ cu 11,2 luni pentru atezolizumab şi, respectiv, docetaxel), cât şi la pacienţii cu NSCLC scuamos (RR de 0,73, IÎ 95%: 0,54; 0,98; SG mediană de 8,9 comparativ cu 7,7 luni pentru atezolizumab şi, respectiv, docetaxel). Îmbunătăţirea observată a SG a fost demonstrată în mod consecvent în subgrupurile de pacienţi, incluzând pacienţii cu metastaze cerebrale la momentul iniţial (RR de 0,54, IÎ 95%: 0,31; 0,94; SG mediană de 20,1 luni pentru atezolizumab, comparativ cu 11,9 luni pentru docetaxel) şi pacienţi care nu au fost niciodată fumători (RR de 0,71, IÎ 95%: 0,47; 1,08;
SG mediană de 16,3 vs. 12,6 luni pentru atezolizumab şi, respectiv, docetaxel). Cu toate acestea, pacienţii cu mutaţii EGFR nu au prezentat îmbunătăţiri ale SG cu atezolizumab, comparativ cu docetaxel (RR de 1,24, IÎ 95%: 0,71; 2,18; SG mediană de 10,5 comparativ cu 16,2 luni pentru atezolizumab şi, respectiv, docetaxel).
S-a observat prelungirea intervalului de timp până la deteriorare în ceea ce priveşte durerea toracică raportată de pacient, evaluată cu ajutorul EORTC QLQ-LC13 în cazul tratamentului cu atezolizumab comparativ cu docetaxel (RR de 0,71, IÎ 95%: 0,49; 1,05; mediana nefiind atinsă în niciunul dintre braţele de tratament). Timpul până la deterioare în ceea ce priveşte alte simptome ale cancerului pulmonar (de exemplu tuse, dispnee şi durere la nivelul braţului/umărului) evaluat cu ajutorul EORTC QLQ-LC13 a fost similar pentru atezolizumab şi docetaxel. Aceste rezultate trebuie interpretate cu precauţie datorită design-ului de studiu deschis.
POPLAR (GO28753): Studiu clinic de fază II, randomizat la pacienţi cu NSCLC local avansat sau metastazat cărora li s-a administrat anterior chimioterapie.
Un studiu clinic de fază II, multicentric, internaţional, randomizat, deschis, controlat, POPLAR, a fost efectuat la pacienţi cu NSCLC local avansat sau metastazat care au avut progresie pe durata sau ulterior unei scheme terapeutice pe bază de săruri de platină, indiferent de expresia PD-L1. Criteriul principal de evaluare a eficacităţii a fost supravieţuirea generală. În total au fost randomizaţi 287 pacienţi în raport de 1:1 pentru a fi trataţi fie cu atezolizumab (1200 mg prin perfuzie intravenoasă la interval de 3 săptămâni, până la pierderea beneficiului clinic) sau docetaxel (75 mg/m2 prin perfuzie
intravenoasă în ziua 1 a fiecărui ciclu de 3 săptămâni, până la progresia bolii). Randomizarea a fost stratificată în funcţie de statusul expresiei PD-L1 la nivelul CI, în funcţie de numărul schemelor anterioare de chimioterapie şi de profilul histologic. O analiză actualizată cu un total de 200 decese observate şi o durată mediană a urmăririi supravieţuirii de 22 luni a indicat o SG mediană de 12,6 luni la pacienţii trataţi cu atezolizumab, comparativ cu 9,7 luni la pacienţii trataţi cu docetaxel (RR de 0,69,
IÎ 95%: 0,52; 0,92). RRO a fost de 15,3% comparativ cu 14,7% şi DR mediană a fost de 18,6 luni pentru atezolizumab, comparativ cu 7,2 luni pentru docetaxel.
Copii şi adolescenţi
Agenţia Europeană pentru Medicamente a suspendat temporar obligaţia de depunere a rezultatelor studiilor efectuate cu Tecentriq la toate subgrupele de copii şi adolescenţi în neoplasmele maligne (cu excepţia tumorilor sistemului nervos central, neoplasmelor ţesutului hematopoietic sau limfoid) (vezi pct. 4.2 pentru informaţii privind utilizarea la copii şi adolescenţi)
5.2 Proprietăţi farmacocinetice
Expunerea la atezolizumab a crescut direct proporţional cu doza pentru intervalul de doze cuprins între 1 mg/kg şi 20 mg/kg, incluzând doza fixă de 1200 mg administrată la interval de 3 săptămâni. O analiză populaţională care a inclus 472 pacienţi a descris farmacocinetica atezolizumab pentru intervalul de doze: 1 până la 20 mg/kg, cu un model de dispoziţie liniară cu două compartimente, cu eliminare de ordinul întâi. O analiză de farmacocinetică populaţională sugerează că starea de echilibru este obţinută după 6 până la 9 săptămâni (2 până la 3 cicluri) de administrare în doze repetate. Acumularea sistemică în ceea ce priveşte aria de sub curba concentraţiei plasmatice în funcţie de timp, concentraţia plasmatică maximă şi concentraţia plasmatică minimă a fost de 1,91, 1,46 şi, respectiv, 2,75 ori.
Absorbţie
Atezolizumab se administrează sub formă de perfuzie intravenoasă. Nu s-au efectuat studii privind alte căi de administrare.
Distribuţie
O analiză de farmacocinetică populaţională indică faptul că, la pacientul tipic, volumul de distribuţie în compartimentul central este de 3,28 l şi volumul la starea de echilibru este de 6,91 l.
Metabolizare
Metabolizarea atezolizumab nu a fost studiată direct. Anticorpii sunt eliminaţi predominant prin catabolism.
Eliminare
Conform unei analize farmacocinetice populaţionale, clearance-ul atezolizumab este de 0,200 l/zi şi timpul de înjumătăţire plasmatică terminal este, în mod obişnuit, de 27 zile.
Grupe speciale de pacienţi
Pe baza analizei FC populaţionale şi a analizei relaţiei expunere-răspuns, vârsta (21-89 ani), regiunea, etnia, insuficienţa renală, insuficienţa hepatică uşoară, nivelul expresiei PD-L1 sau statusul de performanţă ECOG nu au niciun efect asupra farmacocineticii atezolizumab. Greutatea corporală, sexul, statusul pozitiv al AAT, concentraţiile albuminei şi încărcătura tumorală au un efect semnificativ statistic, dar nu relevant din punct de vedere clinic asupra farmacocineticii atezolizumab. Nu se recomandă ajustarea dozei.
Vârstnici
Nu s-au efectuat studii cu atezolizumab la pacienţi vârstnici. Efectul vârstei asupra farmacocineticii atezolizumab a fost evaluat într-o analiză de farmacocinetică populaţională. Vârsta nu a fost identificată ca fiind o covariabilă semnificativă cu influenţă asupra farmacocineticii pe baza intervalului de vârstă al pacienţilor de 21-89 ani (n=472) şi vârsta mediană de 62 ani. Nu a fost observată o diferenţă semnificativă clinic a farmacocineticii atezolizumab între pacienţii cu vârsta < 65 ani (n=274), pacienţii cu vârsta cuprinsă între 65−75 ani (n=152) şi pacienţii cu vârsta >75 ani (n=46) (vezi pct. 4.2).
Copii şi adolescenţi
Nu s-au efectuat studii pentru a investiga farmacocinetica atezolizumab la copii sau adolescenţi.
Insuficienţă renală
Nu s-au efectuat studii speciale cu atezolizumab la pacienţi cu insuficienţă renală. Într-o analiză de farmacocinetică populaţională, nu au fost observate diferenţe importante clinic în ceea ce priveşte clearance-ul atezolizumab la pacienţi cu insuficienţă renală uşoară (rata de filtrare glomerulară estimată [RFGe] 60 – 89 ml/min/1,73 m2 ; n=208) sau moderată (RFGe 30 – 59 ml/min/1,73 m2 ; n=116), comparativ cu pacienţii cu funcţie renală normală (RFGe mai mare sau egală cu 90 ml/min/1,73 m2; n=140). Doar câţiva pacienţi au avut insuficienţă renală severă (RFGe 15 – 29 ml/min/1,73 m2; n=8) (vezi pct. 4.2). Efectul insuficienţei renale severe asupra farmacocineticii atezolizumab nu este cunoscut.
Insuficienţă hepatică
Nu s-au efectuat studii speciale cu atezolizumab la pacienţi cu insuficienţă hepatică. Într-o analiză de farmacocinetică populaţională, nu au fost observate diferenţe importante clinic în ceea ce priveşte clearance-ul atezolizumab la pacienţi cu insuficienţă hepatică uşoară (bilirubina ≤ LSVN şi AST > LSVN sau bilirubina > 1,0 × până la 1,5 × LSVN şi orice valoare a AST, n= 71) şi funcţia hepatică normală (bilirubina şi AST ≤ LSVN, n= 401). Nu sunt disponibile date la pacienţi cu insuficienţă hepatică moderată sau severă. Insuficienţa hepatică a fost definită pe baza criteriilor de disfuncţie hepatică ale Institutului Naţional de Cancer (NCI) (vezi pct. 4.2). Efectul insuficienţei hepatice moderate sau severe (bilirubina > 1,5 × până la 3 × LSVN şi orice valori ale AST sau bilirubina ≥ 3 × LSVN şi orice valori ale AST) asupra farmacocineticii atezolizumab nu este cunoscut.
5.3 Date preclinice de siguranţă
Carcinogenitate
Nu s-au efectuat studii privind carcinogenitatea pentru a stabili potenţialul carcinogen al atezolizumab.
Mutagenitate
Nu s-au efectuat studii privind mutagenitatea pentru a stabili potenţialul mutagen al atezolizumab. Cu toate acestea, nu este de aşteptat ca anticorpii monoclonali să modifice ADN-ul sau cromozomii.
Fertilitate
Nu s-au efectuat studii cu atezolizumab privind fertilitatea; cu toate acestea, evaluarea organelor de reproducere masculine şi feminine a fost inclusă în studiul privind toxicitatea cronică la maimuţe cynomolgus. Administrarea săptămânală a atezolizumab la femelele de maimuţă la o valoare estimată a ASC de 6 ori mai mare decât ASC la pacienţii cărora li s-a administrat doza recomandată a provocat un model de cicluri menstruale neregulate şi absenţa corpului luteal la nivelul ovarelor, care au fost reversibile. Nu a existat nici un efect asupra organelor de reproducere masculine.
Teratogenitate
Nu s-au efectuat studii cu atezolizumab privind teratogenitatea şi asupra funcţiei de reproducere la animale. Studiile la animale au evidenţiat că inhibarea pe calea PD-L1/PD-1 poate duce la respingerea mediată imun a fetusului aflat în dezvoltare, provocând deces fetal. Administrarea atezolizumab poate avea efecte dăunătoare asupra fătului, incluzând letalitate embrio-fetală.
6. PROPRIETĂŢI FARMACEUTICE
6.1 Lista excipienţilor
L-histidină
Acid acetic glacial
Sucroză
Polisorbat 20
Apă pentru preparate injectabile
6.2 Incompatibilităţi
Acest medicament nu trebuie amestecat cu alte medicamente, cu excepţia celor menţionate la pct. 6.6.
6.3 Perioada de valabilitate
Flacon nedeschis
2 ani
Soluţie diluată
Stabilitatea chimică şi fizică în uz a fost demonstrată pentru maxim 24 ore, la temperaturi de 2-8°C sau 8 ore la temperatura camerei (temperaturi ≤ 30°C) din momentul pregătirii.
Din punct de vedere microbiologic, soluţia preparată pentru perfuzare trebuie utilizată imediat. Dacă nu este utilizată imediat, perioada de timp şi condiţiile de păstrare înainte de utilizare sunt responsabilitatea utilizatorului.
6.4 Precauţii speciale pentru păstrare
A se păstra la frigider (2°C-8°C).
A nu se congela.
A se ţine flaconul în cutie pentru a fi protejat de lumină.
Pentru condiţiile de păstrare ale medicamentului după diluare, vezi pct. 6.3.
6.5 Natura şi conţinutul ambalajului
Flacon din sticlă de tip I cu dop din cauciuc butilic, conţinând 20 ml soluţie.
Cutie cu un flacon.
6.6 Precauţii speciale pentru eliminarea reziduurilor <şi alte instrucţiuni de manipulare>
Tecentriq nu conţine niciun conservat antimicrobian şi trebuie pregătit de către un profesionist în domeniul sănătăţii, utilizând o tehnică aseptică.
A nu se agita.
Instrucţiuni privind diluarea
Se extrag din flacon douăzeci ml Tecentriq concentrat şi se diluează într-o pungă de perfuzie din PVC, polietilenă (PE) sau poliolefină cu capacitatea de 250 ml conţinând soluţie de clorură de sodiu 9 mg/ml (0,9%) pentru preparate injectabile. După diluare, un ml din soluţie trebuie să conţină aproximativ 4,4 mg Tecentriq (1200 mg/270 ml). Punga trebuie întoarsă uşor pentru a amesteca soluţia, astfel încât să se evite formarea spumei. După pregătirea soluţiei perfuzabile, aceasta trebuie administrată imediat
(vezi pct. 6.3).
Medicamentele cu administrare parenterală trebuie inspectate vizual pentru a depista prezenţa particulelor şi a modificărilor de culoare înainte de administrare. Dacă se observă prezenţa particulelor sau a modificărilor de culoare, soluţia nu trebuie utilizată.
Nu au fost observate incompatibilităţi între Tecentriq şi pungile pentru perfuzie intravenoasă din clorură de polivinil (PVC), polietilenă (PE) sau poliolefină (PO) ale căror suprafeţe vin în contact cu medicamentul. În plus, nu au fost observate incompatibilităţi între membranele filtrului încorporat compus din polietersulfonă sau polisulfonă şi seturile de perfuzie şi alte instrumente utilizate pentru perfuzare şi compuse din PVC, PE, polibutadienă sau polieteruretan. Utilizarea membranelor filtrului încorporat este opţională.
Eliminarea
Eliminarea Tecentriq în mediul înconjurător trebuie redusă la minim. Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW
Marea Britanie
8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
EU/1/17/1220/001
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
10. DATA REVIZUIRII TEXTULUI
Informaţii detaliate privind acest medicament sunt disponibile pe site-ul Agenţiei Europene pentru Medicamente http://www.ema.europa.eu.